
 木盒主机
木盒主机2022年6月7日,来自多伦多大学计算机科学系的Daniel Flam-Shepherd等人在Nat Commun发表研究工作,研究为分子的深度生成模型引入了三个复杂的建模任务来测试化学语言模型的能力,结果显示语言模型是可以学习任何复杂分子分布的非常强大的生成模型。

摘要
分子的深度生成模型已经得到了极大的普及,这些模型在相关的数据集上进行训练,被用来搜索整个化学空间。用于逆向设计新型功能化合物的生成模型的价值,取决于其学习分子分布的能力。最简单的例子是一种语言模型,它采用循环神经网络的形式,并用字符串表征生成分子。
在这项工作中,我们研究了简单语言模型学习更复杂的分子分布的能力。为此,我们编译了更大、更复杂的分子分布,引入了几个具有挑战性的生成性建模任务,并评估了语言模型在每个任务上的能力。
结果表明,语言模型是强大的生成模型,能够熟练地学习复杂的分子分布。语言模型可以准确地生成:ZINC15中得分最高的惩罚性LogP分子的分布,多模式的分子分布以及PubChem中最大的分子。这些结果突出了一些最流行的和最近的图生成模型的局限性,其中许多模型无法扩展到这些分子分布。
引言
对化学空间的有效探索在药物和材料发现方面有许多应用。然而,探索工作只探究了可合成的化学空间的一个很小的子集,因此开发新的工具是至关重要的。鉴于人工智能在其他具有挑战性的科学问题 (如蛋白质结构预测) 上的成功,人工智能的兴起可能为揭开化学世界的神秘面纱提供了方法。
最近,深度生成模型已经成为应对这一巨大挑战的最有希望的工具之一。这些模型在化学空间的相关子集上进行训练,并能生成与其训练数据类似的新分子。它们学习训练分布并生成有效的、类似的分子的能力,对于在下游应用 (如功能化合物的逆向设计) 中取得成功非常重要。
语言模型可用于生成用于药物发现的分子库,或内置到变异自动编码器 (VAE),其中贝叶斯优化可用于通过模型的潜在空间搜索类药分子。其他模型使用图神经网络按顺序将分子生成为图或一次性生成整个分子。最流行的两个:CGAVE和JTVAE可以被直接约束以强制执行化合价限制。
语言模型已被广泛应用,研究人员将其用于基于配体的从头设计。最近使用的一些语言模型案例包括:靶向受天然产物启发的类视黄醇X受体调节剂,设计肝脏X受体激动剂,从基因表达特征生成类似hit的分子等。其他研究强调了语言模型在低数据系统中的能力,并可通过数据增强来提高性能。
最初,SMILES字符串表征法的脆弱性意味着一个字符就可能导致无效的分子。这个问题在很大程度上已经被更强大的分子字符串表征法所解决。此外,随着训练方法的改进,基于RNN的深度生成模型一直使用SMILES生成高比例的有效分子。尚未研究的一个领域是语言模型和生成模型生成更大更复杂的分子、或从大小和结构范围更大的化学空间中生成分子的能力。这是有益的,因为人们对更大更复杂的分子用于治疗的兴趣增加了。
为了测试语言模型的能力,我们通过构建比标准数据集更复杂的分子训练集,制定了一系列具有挑战性的生成性建模任务。特别是,我们关注语言模型学习目标数据集的分布属性的能力。我们在所有的任务上训练语言模型,并以许多其他图生成模型为基准。结果表明,语言模型是强大的生成模型,可以比大多数图生成模型更好地学习复杂的分子分布。
结果
我们定义了三个任务,生成:(1) 惩罚性LogP3高分的分子分布 (图1a, d), (2) 分子的多模式分布 (图1b, e),以及 (3) PubChem中最大的分子 (图1c, e)。必然地,每个不同的生成性建模任务都是通过学习从数据集中的分子分布中生成来定义的。我们使用较大数据库的相关子集建立了三个数据集。
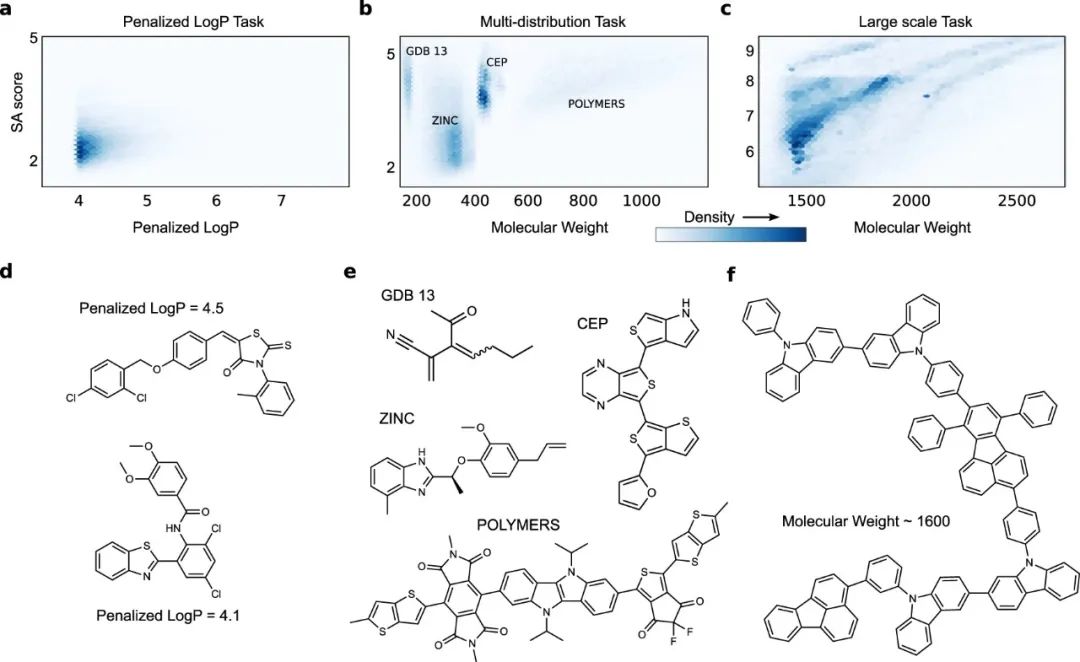
图1:生成式建模任务
a-c 定义了三个复杂分子生成式建模任务的分子分布。a 惩罚性LogP任务中训练数据的惩罚性LogP与SA得分的分布。b 多分布任务的训练数据中不同权重分子的四种模式。c 大规模任务的分子量训练分布。d-f 每个生成式建模任务的训练数据中的分子示例。d 惩罚性LogP任务,e 多分布任务。f 大规模任务。
表1是所有数据集与两个标准数据集Zinc和Moses的原子和环数的一些汇总统计。所有的任务都涉及有更多的子结构的更大的分子,并且每个分子包含更大范围的原子和环数。
表1 所有三个任务的数据集与标准数据集的比较统计
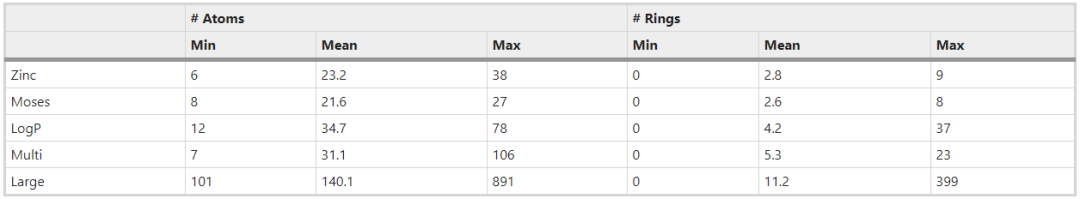
对于每个任务,我们通过绘制训练分子属性的分布以及语言模型和图形模型学习的分布来评估性能。我们对训练分子使用直方图,并通过调整其带宽参数 (bandwidth parameter) 对其进行高斯核密度估计。我们使用相同的带宽参数为所有模型的分子属性绘制KDE。
从所有的模型中,我们最初生成1万个分子,计算它们的属性,并使用它们来生成所有的图和指标。此外,为了公平地比较所学到的分布,我们在去除重复的和训练过的分子后,从所有模型中使用相同数量的生成分子。
为了定量评估任何模型学习其训练分布的能力,我们计算了生成分子和训练分子的属性值之间的Wasserstein距离。我们还计算了不同训练分子样本之间的Wasserstein距离,以确定一个最理想的基线,我们可以将其作为一个预言机 (oracle) 来比较。
对于分子特性,我们考虑:药物相似性的定量估计 (QED),合成可及性得分 (SA),辛醇-水分配系数 (Log P),精确分子量 (MW),伯特兹复杂性 (BCT),天然产物相似性 (NP)。我们还使用标准指标,如有效性、独特性、新颖性,来评估模型生成不同于训练数据的各种真实分子的能力。
对于模型,我们主要考虑的是使用具有长短期记忆的递归神经网络的化学语言模型,并在SMILES (SM-RNN) 或SELFIES (SF-RNN) 上训练。我们还训练两个最流行的深度图生成模型:结点树变异自动编码器 (JTVAE) 和约束图变异自动编码器 (CGVAE)。
惩罚性LogP任务
对于第一项任务,我们考虑了最广泛使用的搜索化学空间的基准评估之一,即惩罚性LogP任务,寻找具有高LogP的分子,并以可合成性和不切实际的环作为惩罚。我们考虑这个任务的生成模型版本,目标是学习具有高惩罚性LogP分数的分子的分布。寻找具有良好分数 (3.0以上) 的单个分子是一个标准的挑战,但是学习从化学空间的这一部分直接生成,使模型产生的每个分子都具有较高的惩罚性LogP,则增加了另一种程度的难度。为此,我们通过在ZINC15数据库中筛选出惩罚性LogP值超过4.0的分子来建立一个训练数据集。
经过筛选,ZINC中得分最高的分子大约为160K个分子,用于该任务的训练数据。
所有模型的训练结果都显示在图2和图3中。语言模型的表现比图形模型好,SELFIES RNN产生的结果与图2a中的训练分布稍微接近。CGVAE和JTVAE学习产生了大量的分子,其惩罚后的LogP分数大大低于最低训练分数。
在图2b-d中,我们看到JTVAE和CGVAE学习产生了更多比训练数据更大的SA分数的分子,同样,我们看到所有模型都学习了训练数据中LogP的主要模式,但是RNN产生了更接近的分布--对于QED也可以看到类似的结果。这些结果延续到了定量指标上,两个RNN都取得了比CGVAE和JTVAE更低的Wasserstein距离指标 (表2),SMILES RNN最接近TRAIN预言机。
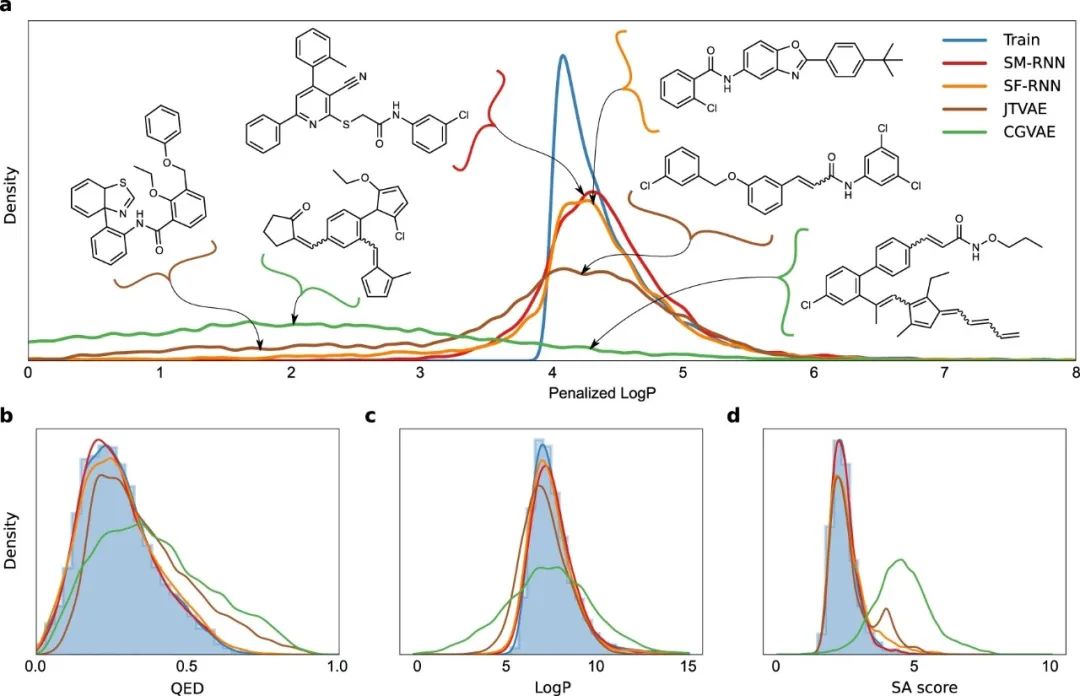
图2:惩罚性的LogP任务I
a 使用在SMILES上训练的SM-RNN、在SELFIES上训练的SF-RNN和图形模型:CGVAE和JTVAE的训练数据 (TRAIN) 中分子的惩罚LogP分数的绘图分布。
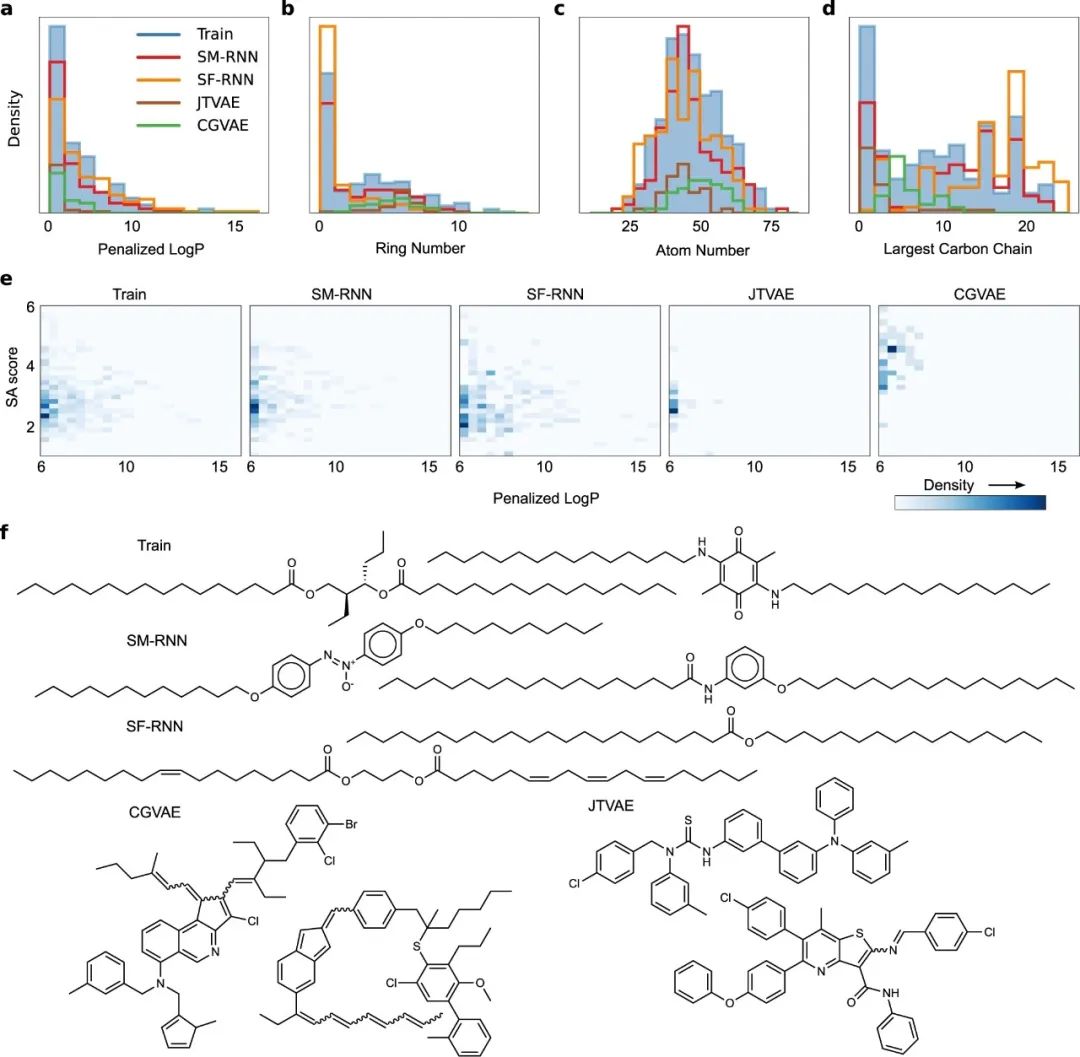
图3:惩罚性LogP任务II
a-d 所有模型生成的分子或训练数据中惩罚性LogP≥6.0的分子的惩罚性LogP、原子数、环数和最大碳链长度 (均为每个分子) 的直方图。f 所有模型生成的少数分子或训练数据中惩罚性LogP≥6.0的分子的直方图。
表2 所有三个任务中来自训练数据和由模型生成的分子之间的LogP、SA、QED、MW、BT和NP的Wasserstein距离指标。
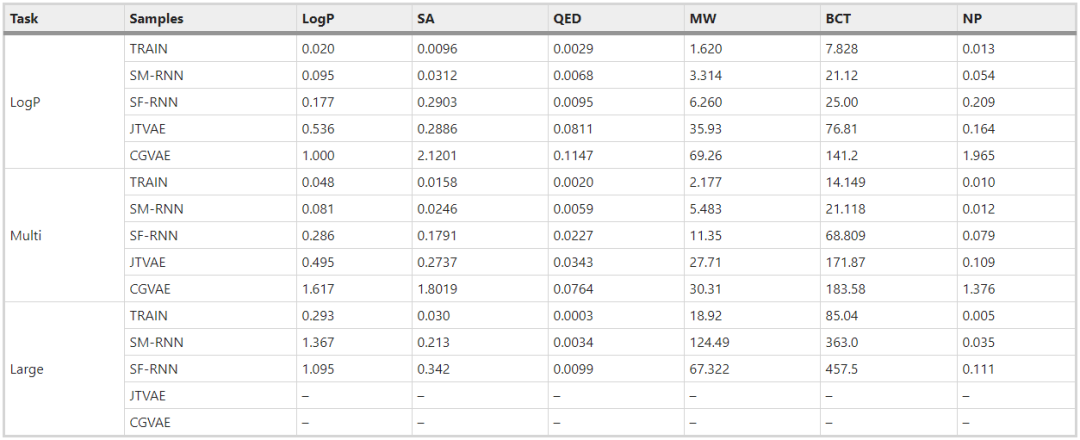
我们进一步研究训练数据中数值超过6.0的最高惩罚LogP区域--训练分布的微妙尾部。在2d分布中 (图3e),很明显,两个RNN都学习了训练数据的这个微妙的方面,而图模型几乎完全忽略了它,只学习了更接近主模式的分子。特别是,CGVAE学习的分子的SA分数比训练数据大。
此外,训练数据中惩罚性LogP得分最高的分子通常含有很长的碳链和较少的环 (图3b,d) --RNNs能够发现这一点。这在模型产生的样本中非常明显,图3f显示了一些样本,RNNs产生的大部分分子都有长碳链,而CGVAE和JTVAE产生的分子有很多环,其惩罚性LogP分数接近6.0。在图3a-d的直方图中,语言模型学习的分布与训练分布接近。
总的来说,语言模型可以学习具有高惩罚性LogP分数的分子分布,比图形模型更好。
多分布任务
对于下一个任务,我们通过结合以下子集创建了一个数据集。(1) 分子量 (MW) ≤185的GDB13分子,(2) 185≤MW≤425的ZINC分子,(3) 460≤MW≤600的Harvard CEP (clean energy project) 分子,以及 (4) MW>600的POLYMERS分子。训练分布有四个模式 (图1b、e和4a)。
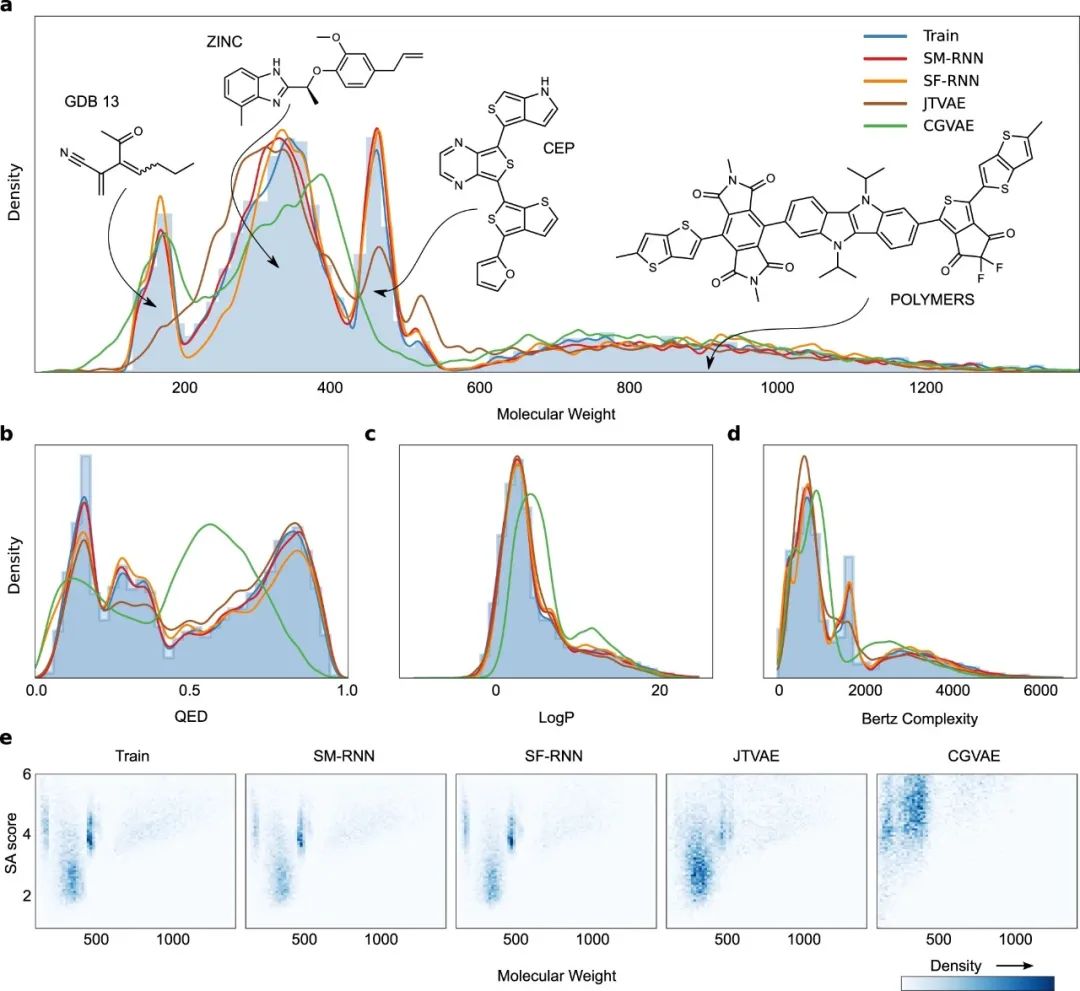
图4:多分布任务
a 训练分子的直方图和KDE,以及从所有模式产生的分子的KDE。b-d 训练分子的QED、LogP和SA分数的直方图和KDE以及所有模型产生的分子的KDES。e 所有模型生成的训练分子和分子的分子量和 SA 分数的二维直方图。
在多分布任务中,两个RNN模型都能很好地捕捉数据分布,并学习训练分布中的每个模式 (图4a)。另一方面,JTVAE完全错过了GDB13的第一个模式,然后对ZINC和CEP的学习很差。同样,CGVAE也学习了GDB13,但低估了ZINC,并完全错过了CEP的模式。更多的证据表明,RNN模型更紧密地学习了训练分布,这在图4e中很明显,CGVAE和JTVAE几乎没有区分出主要模式。此外,RNN模型产生的分子与训练数据更相似 (补充表4)。尽管如此,所有的模型--除了CGVAE,都能捕捉到QED、SA得分和Bertz复杂度的训练分布 (图4b-d)。最后,在表2中,在SMILES上训练的RNN具有最低的Wasserstein度量,其次是SELFIES RNN,然后是JTVAE和CGVAE。
大规模的任务
最后一项生成模型任务,涉及测试深度生成模型学习大分子的能力,即与使用SMILES/SELFIES字符串表征或图形的分子生成模型相关的最大可能的分子。为此,我们转向PubChem,筛选出有100多个重原子的最大分子,产生了大约30万个分子。它们的分子量范围也很广,从1250到5000,但大多数分子属于1250-2000范围 (图1c)。
这项任务对图模型来说是最具挑战性的,两者都未能训练并且完全无法学习训练数据。特别是,JTVAE的树状分解算法应用于训练数据产生了一个固定的词汇表,即11000个子结构。然而,两个RNN模型都能够学习生成与训练数据一样大、一样多的分子。训练分子对应于很长的SMILES和SELFIES字符串表征,在这种情况下,SELFIES字符串提供了额外的优势,SELFIES RNN可以更接近于数据分布 (图5a)。特别是,用SMILES语法学习有效的分子大大增加了难度,因为这些分子有更多的字符需要生成,模型犯错并产生无效字符串的概率更高。相比之下,生成的SELFIES字符串永远不会是无效的。有趣的是,即使RNN模型生成的分子不在分布范围内,而且比训练分子小得多--它们仍然有类似的子结构,并与训练分子相似 (图5a)。
此外,训练分子似乎分为两种模式,即LogP值较低和较高的分子 (图5b):生物大分子定义较低的模式,而具有更多环和更长碳链的分子定义较高的LogP模式(更多的例子分子可以在补充图8中看到)。RNN模型都能够学习训练分布的双模性质。
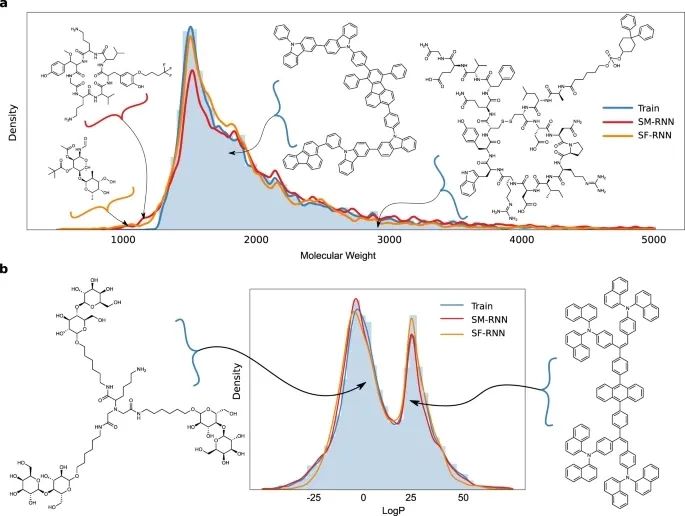
图5:大规模的任务I
a 训练分子的直方图和KDE以及由RNN生成的分子量的KDE。图中左边显示了两个由RNN生成的分子量低于训练分子的分子。此外,分子量分布的模式和尾部的两个训练分子显示在右边。b 训练分子的直方图和对数值的KDE以及由RNNs生成的分子的对数值的KDE。在图的两边,对于LogP分布中的每个模式,我们显示了训练数据中的一个分子。
训练数据有各种不同的分子和子结构,在图6a中,RNN模型充分地学习了训练分子中出现的子结构的分布。特别是数量的分布:每个分子中的片段、单原子片段以及单个、稠环和氨基酸片段。随着训练分子越来越大并且出现的次数越来越少,两个RNN模型仍能学会生成这些分子(图5a,当分子量>3000时)。
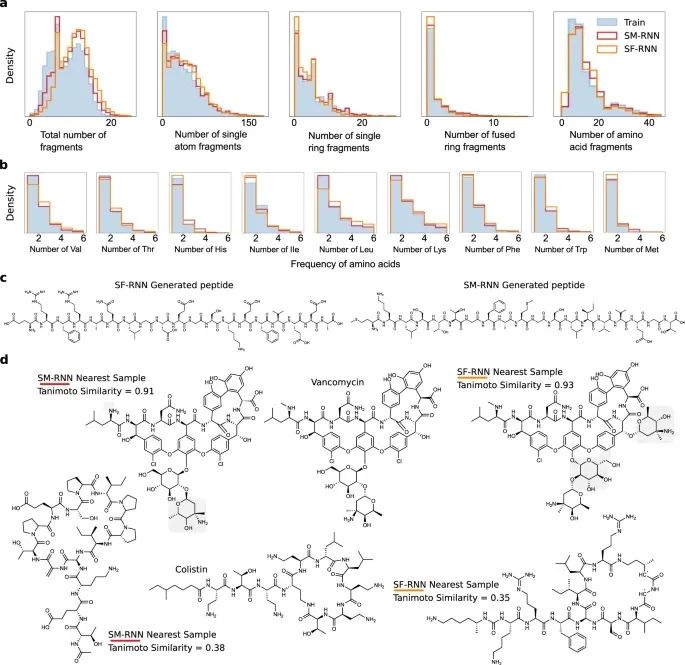
图6:大规模的任务II
a 由RNN模型或训练数据生成的分子中的片段#、单原子片段#、单环片段#、稠环片段#、氨基酸片段#(均为每个分子)的直方图。b 由RNN模型或训练数据生成的每个分子中特定氨基酸数量的直方图。c 由SM-RNN-MKLSTTGFAMGSLIVVEGT (右) 和SF-RNN-ERFRAQLGDEGSKEFVEEA(左)生成的多肽。d 由SF-RNN和SM-RNN生成的分子在Tanimoto相似度上与科利司汀和万古霉素最接近。浅灰色的阴影区域突出了与万古霉素的差异。
这个任务中的数据集包含了PubChem中出现的一些肽和环状肽,我们直观地分析了RNNs的样本,看它们是否能够保留主链结构和天然氨基酸。我们发现,RNNs经常对主链的片段进行采样,这些主链通常是不连接的--与其他原子、键和结构断开。此外,通常这些链有来自主要氨基酸残基的标准侧链,但也有其他非典型的侧链出现。在图6c中,我们展示了两个由SM-RNN和SF-RNN生成的肽的例子。
虽然有很多例子,这两个模型都没有保留骨架,幻想出奇怪的侧链,但很有可能,如果完全在相关的肽上进行训练,该模型就可以用于肽的设计。甚至更进一步,由于这些语言模型不限于生成氨基酸序列,可以用来设计任何模仿肽类结构的生化结构,甚至复制它们的生物行为。这使得它们非常适用于设计修饰过的肽,其他肽的模仿物和复杂的天然产品。唯一的要求是由领域专家来构建特定目标的训练数据集。
我们对RNNs在训练数据中学习生物分子结构的情况进行了额外的研究,在图6b中,我们看到两个RNNs都与必需氨基酸的分布相匹配 (使用子结构搜索发现)。最后,RNNs也有可能被用来设计环状肽。为了强调语言模型在这项任务中的前景,我们展示了由RNNs生成的与大肠杆菌素和万古霉素相似度最高的分子 (图6d)。这项任务的结果表明,语言模型可以用来设计更复杂的生物大分子。
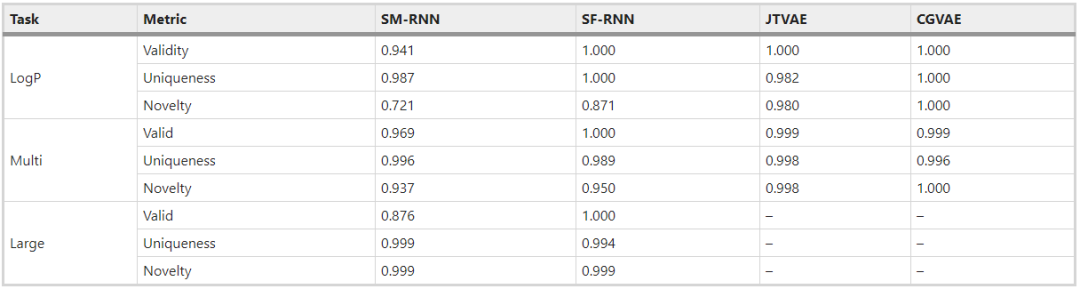
我们还根据文献中的标准指标来评估模型:有效性、独特性和新颖性。使用每个模型为每个任务生成的相同的10K分子,我们计算了参考文献中定义的以下统计数据。并存储在表3中:(1) 有效性:有效分子和生成分子的数量之比; (2) 唯一性:唯一分子(不重复的) 和有效分子的数量之比;(3) 新颖性:训练数据中没有的唯一分子和唯一分子总数之比。
在前两个任务中 (表3),JTVAE和CGVAE有更好的指标,具有非常高的有效性、唯一性和新颖性 (都接近1),这里SMILES和SELFIES RNN表现较差,但SELFIES RNN与它们的性能接近。由于SMILES RNN的语法较差,其指标也较差,但与其他模型相比,并没有实质性的差。
表3 所有模型在每个任务中生成的分子的标准指标有效性、独特性和新颖性
我们还考虑了许多额外的图生成模型基线的所有任务。这些包括一些GANs,一些自回归模型,归一化流和单次模型大多数根本无法扩展,少数有规模的基线-只能处理LogP和多分布的任务,但没有比语言模型表现更好。
讨论
在这项工作中,为了测试化学语言模型的能力,我们为分子的深度生成模型引入了三个复杂的建模任务。语言模型和图基线执行每个任务,这需要学习从具有挑战性的数据集中生成分子。结果表明,语言模型是非常强大、灵活的模型,可以学习各种非常不同的复杂分布,而流行的图基线的能力要差得多。
在SELFIES和SMILES的比较中,SM-RNN和SF-RNN在所有任务中都表现良好,比基线好。我们报告说,SF-RNN在每个任务中都有更好的标准指标 (表3),但SM-RNN有更好的Wasserstein距离指标 (表2)。此外,SF-RNN比SM-RNN有更好的新颖性--这可能意味着SELFIES语法导致语言模型对训练数据集的记忆减少。这也可以帮助解释为什么SF-RNN有更好的标准指标,但比SM-RNN的Wasserstein指标差。此外,数据增强和随机SMILES可以用来提高SM-RNN的新颖性得分。在未来,对SMILES和SELFIES表征在深度生成模型中的使用进行更全面的评估是有价值的。
结果表明,主要的基线图生成模型,JTVAE和CGVAE并不像语言模型那样灵活。对于惩罚性的LogP任务,一个得2分的分子和一个得4分的分子之间的差别往往是非常细微的。有时改变一个碳原子或其他原子就会导致分数大幅下降--这可能解释了为什么CGVAE与主要训练模式严重不匹配。对于多分布任务,JTVAE和CGVAE的困难很明显,但非常容易理解。对于JTVAE来说,它必须学习广泛的树类型:其中许多树类型没有像环一样的大型子结构 (GDB13分子),而其他树类型则完全是环 (CEP和POLYMERS)。对于CGVAE来说,它必须学习广泛的非常不同的生成轨迹--这是很困难的,尤其是在学习过程中它只使用一个样本轨迹。由于同样的原因,这些模型没有能力对PubChem中最大的分子进行训练。
语言模型也比额外的图生成基线表现得更好--它们具有与JTVAE和CGVAE相同的限制。这几乎是意料之中的,因为图生成模型有生成原子和键信息的更困难的任务,而语言模型只需要生成一个单一的序列。鉴于此,语言模型显示出如此灵活的能力是很自然的,而被评估的图生成模型则没有。在分子设计之外,一些图生成模型已经试图扩展到更大的图,但是这些模型还没有被增强到分子中。这里的结果确实突出了这样一个事实:许多广泛使用的图生成模型只为类似药物的小分子而设计,不能扩展到更大更复杂的分子。另一方面,虽然语言模型可以扩展并灵活地生成更大的分子,但图生成模型更具有可解释性,这对药物和材料的发现很重要。
根据所进行的实验,语言模型是学习任何复杂分子分布的非常强大的生成模型,应该看到更广泛的应用。然而,仍有可能看到这些模型的改进,因为这些模型不能考虑其他重要的信息,如分子几何学。此外,我们希望所介绍的分子建模任务和数据集能够促使为这些更大、更复杂的分子建立新的生成模型。
参考资料
Flam-Shepherd, D., Zhu, K. & Aspuru-Guzik, A. Language models can learn complex molecular distributions. Nat Commun 13, 3293 (2022). https://doi.org/10.1038/s41467-022-30839-x
--------- End ---------
未经允许不得转载:木盒主机 » Nat Commun|语言模型可以学习复杂的分子分布
 搬瓦工VPS最新优惠码 搬瓦工最高优惠6.81%优惠码 promo coupon code
搬瓦工VPS最新优惠码 搬瓦工最高优惠6.81%优惠码 promo coupon code  RackNerd:美国VPS 黑五优惠折扣 1核768RAM $10.28/年+神秘盒子 可随机减免金额
RackNerd:美国VPS 黑五优惠折扣 1核768RAM $10.28/年+神秘盒子 可随机减免金额 10G.BIZ【年终钜惠】美国/日本/韩国/香港独立服务器 秒杀仅24起,站群仅需99,三网CN2GIA五折抢购。CERA洛杉矶云服务器仅2.4起
10G.BIZ【年终钜惠】美国/日本/韩国/香港独立服务器 秒杀仅24起,站群仅需99,三网CN2GIA五折抢购。CERA洛杉矶云服务器仅2.4起 2022年RackNerd 美国VPS促销:4TB月流量11.88美元/年,支持支付宝,老优惠$9.89美元/年
2022年RackNerd 美国VPS促销:4TB月流量11.88美元/年,支持支付宝,老优惠$9.89美元/年