
 木盒主机
木盒主机使用TCGAbiolinks包探索最新版TCGA数据库:
新版TCGAbiolinks包学习:批量下载数据
新版TCGAbiolinks包学习:表达矩阵提取(mRNA/lncRNA/counts/tpm/fpkm)
手动下载的TCGA数据也是可以用TCGAbiolinks包整理的
新版TCGAbiolinks包学习:差异分析
新版TCGAbiolinks包学习:富集分析和生存分析
TCGA的maf突变文件不能下载了?直接用TCGAbiolinks包搞定!
TCGAbiolinks的甲基化数据分析
今天学习下TCGAbiolinks包中的可视化函数。
这些图形都可以使用其他R包进行更好看的可视化,平常大家根本不用,不过作为TCGAbiolinks包完整学习的一部分,在这里简单记录一下。。
<code style="margin-left:0">library(TCGAbiolinks) suppressMessages(library(SummarizedExperiment)) </code>
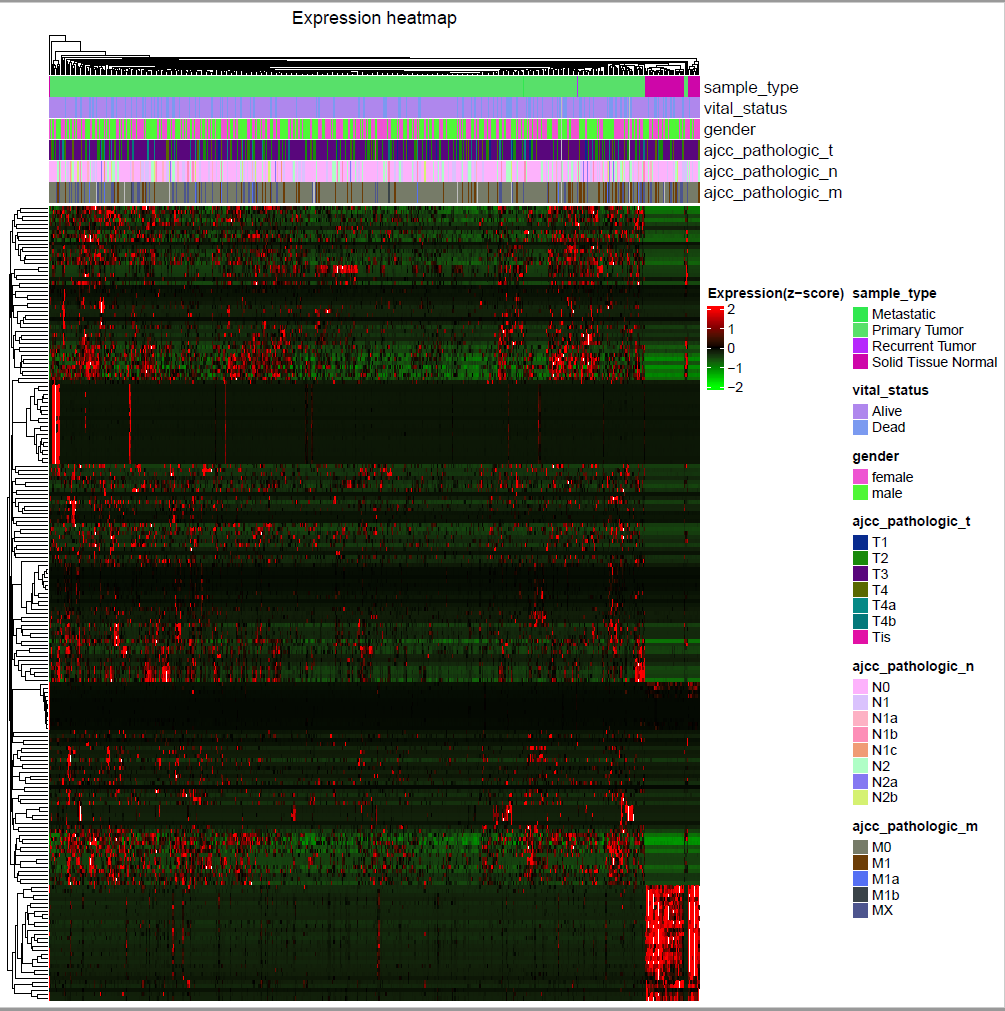
热图
可视化差异基因或者差异甲基化。
使用前面推文中得到的COAD的差异基因演示。
<code style="margin-left:0"># 获取表达矩阵
load("TCGA-mRNA/TCGA-COAD_mRNA.Rdata")
se_mrna <- data[rowData(data)$gene_type == "protein_coding",]
coadMatrix <- assay(se_mrna, "unstranded")
coad_coroutliers <- TCGAanalyze_Preprocessing(se_mrna,cor.cut = 0.7)
## Number of outliers: 0
coadNorm <- TCGAanalyze_Normalization(
tabDF = coad_coroutliers,
geneInfo = geneInfoHT)
## I Need about 127 seconds for this Complete Normalization Upper Quantile [Processing 80k elements /s]
## Step 1 of 4: newSeqExpressionSet ...
## Step 2 of 4: withinLaneNormalization ...
## Step 3 of 4: betweenLaneNormalization ...
## Step 4 of 4: exprs ...
coadFilt <- TCGAanalyze_Filtering(
tabDF = coadNorm,
method = "quantile",
qnt.cut = 0.25
)
# 保存下方便以后使用
#save(coadFilt,file = "./output/coadFilt.rdata")
# 准备差异基因
load(file = "./output/coadDEGs.Rdata")
</code>查看数据:
<code style="margin-left:0">coadFilt[1:4,1:4] ## TCGA-NH-A8F7-06A-31R-A41B-07 TCGA-3L-AA1B-01A-11R-A37K-07 ## ENSG00000000003 15299 7257 ## ENSG00000000005 26 23 ## ENSG00000000419 5139 2058 ## ENSG00000000457 614 734 ## TCGA-4N-A93T-01A-11R-A37K-07 TCGA-4T-AA8H-01A-11R-A41B-07 ## ENSG00000000003 7125 2918 ## ENSG00000000005 67 89 ## ENSG00000000419 2626 844 ## ENSG00000000457 731 323 </code>
查看数据:
<code style="margin-left:0">head(coadDEGs) ## logFC logCPM LR PValue FDR ## ENSG00000000419 1.018334 5.864185 58.75510 1.785702e-14 7.381441e-14 ## ENSG00000000460 1.423716 3.457955 153.99195 2.325351e-35 3.797553e-34 ## ENSG00000000971 -1.052867 4.847858 37.14551 1.096349e-09 3.067592e-09 ## ENSG00000001460 -1.006716 3.531623 170.11301 6.990106e-39 1.377268e-37 ## ENSG00000001497 1.151358 6.086819 114.28071 1.131077e-26 1.106818e-25 ## ENSG00000001617 1.167619 5.505217 51.08214 8.858051e-13 3.195640e-12 ## gene_name gene_type ## ENSG00000000419 DPM1 protein_coding ## ENSG00000000460 C1orf112 protein_coding ## ENSG00000000971 CFH protein_coding ## ENSG00000001460 STPG1 protein_coding ## ENSG00000001497 LAS1L protein_coding ## ENSG00000001617 SEMA3F protein_coding </code>
我们用logFC最大的前500个基因演示:
<code style="margin-left:0">top500 <- coadDEGs[order(abs(coadDEGs$logFC),decreasing =T),][1:500,] </code>
准备热图需要的表达矩阵:
<code style="margin-left:0">heat.df <- coadFilt[rownames(coadFilt) %in% rownames(top500),] dim(heat.df) </code>
准备热图需要的样本信息,必须有一列和表达矩阵的列名相同:
<code style="margin-left:0">coldata <- colData(data)
dim(coldata)
## [1] 521 107
coldata.df <- as.data.frame(subset(coldata, select=c("barcode","sample_type","vital_status","gender",
"ajcc_pathologic_t","ajcc_pathologic_n",
"ajcc_pathologic_m")))
head(coldata.df)
## barcode sample_type
## TCGA-A6-5664-01A-21R-1839-07 TCGA-A6-5664-01A-21R-1839-07 Primary Tumor
## TCGA-D5-6530-01A-11R-1723-07 TCGA-D5-6530-01A-11R-1723-07 Primary Tumor
## TCGA-AA-3556-01A-01R-0821-07 TCGA-AA-3556-01A-01R-0821-07 Primary Tumor
## TCGA-AA-3660-11A-01R-1723-07 TCGA-AA-3660-11A-01R-1723-07 Solid Tissue Normal
## TCGA-AA-3818-01A-01R-0905-07 TCGA-AA-3818-01A-01R-0905-07 Primary Tumor
## TCGA-AA-3660-01A-01R-1723-07 TCGA-AA-3660-01A-01R-1723-07 Primary Tumor
## vital_status gender ajcc_pathologic_t
## TCGA-A6-5664-01A-21R-1839-07 Alive male T4a
## TCGA-D5-6530-01A-11R-1723-07 Alive male T2
## TCGA-AA-3556-01A-01R-0821-07 Alive male T2
## TCGA-AA-3660-11A-01R-1723-07 Alive female T3
## TCGA-AA-3818-01A-01R-0905-07 Dead female T3
## TCGA-AA-3660-01A-01R-1723-07 Alive female T3
## ajcc_pathologic_n ajcc_pathologic_m
## TCGA-A6-5664-01A-21R-1839-07 N2a MX
## TCGA-D5-6530-01A-11R-1723-07 N0 M0
## TCGA-AA-3556-01A-01R-0821-07 N0 M0
## TCGA-AA-3660-11A-01R-1723-07 N0 M0
## TCGA-AA-3818-01A-01R-0905-07 N0 M0
## TCGA-AA-3660-01A-01R-1723-07 N0 M0
</code>然后使用TCGAvisualize_Heatmap函数画热图,其实也是complexheatmap的包装:
<code style="margin-left:0">TCGAvisualize_Heatmap(data = heat.df,
col.metadata = coldata.df,
cluster_rows = T,
cluster_columns = T,
scale = "row",
extremes = seq(-2,2,1),
color.levels = colorRampPalette(c("green", "black", "red"))(n = 5)
)
</code>会在当前目录生成一张热图:
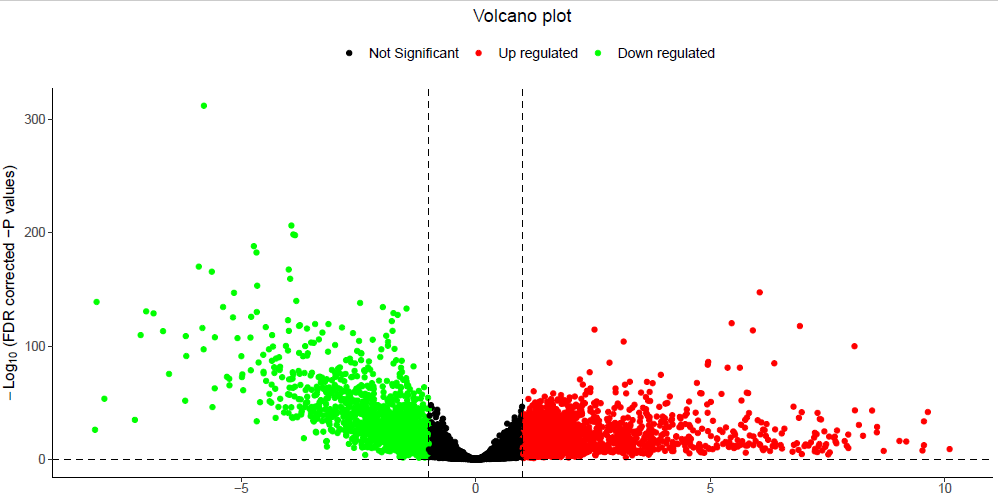
火山图
使用所有基因的差异信息。
<code style="margin-left:0">load(file = "./output/coadDEGsAll.Rdata")
TCGAVisualize_volcano(x=coadDEGAll$logFC,
y=coadDEGAll$FDR, # 纵坐标会自动变成-log10
x.cut = c(-1,1),
y.cut = 2
)
</code>会在当前目录下保存火山图,这个图纵坐标太大了!这样发文章是不太行的哦!
火山图
PCA图
<code style="margin-left:0">rm(list = ls()) load(file = "./output/coadFilt.rdata") load(file = "./output/coadDEGsAll.Rdata") </code>
定义下样本类型:
<code style="margin-left:0"># normal
group1 <- TCGAquery_SampleTypes(colnames(coadFilt), typesample = c("NT"))
# tumor
group2 <- setdiff(colnames(coadFilt), group1)
</code>需要获得一个差异基因table:
<code style="margin-left:0"># DEGs table with expression values in normal and tumor samples
coadDEGsFiltLevel <- TCGAanalyze_LevelTab(
FC_FDR_table_mRNA = coadDEGAll,
typeCond1 = "Normal",
typeCond2 = "Tumor",
TableCond1 = coadFilt[,group1],
TableCond2 = coadFilt[,group2]
)
head(coadDEGsFiltLevel)
## mRNA logFC FDR Delta Normal
## ENSG00000161016 ENSG00000161016 1.0634492 4.608668e-12 74483.67 70039.71
## ENSG00000167658 ENSG00000167658 0.4903969 2.389951e-05 65654.67 133880.68
## ENSG00000137154 ENSG00000137154 0.6959504 2.038518e-08 62058.29 89170.56
## ENSG00000089157 ENSG00000089157 0.7335765 1.574363e-09 55289.82 75370.22
## ENSG00000108821 ENSG00000108821 2.7959748 6.188722e-22 55126.53 19716.39
## ENSG00000111640 ENSG00000111640 0.9146113 1.156871e-12 54971.11 60103.24
## Tumor start end
## ENSG00000161016 99007.33 144789765 144792587
## ENSG00000167658 126985.97 3976056 3985463
## ENSG00000137154 103264.10 19375715 19380236
## ENSG00000089157 86214.63 120196699 120201235
## ENSG00000108821 105676.55 50184101 50201632
## ENSG00000111640 74969.70 6534512 6538374
</code>PCA分析并画图:
<code style="margin-left:0">pca <- TCGAvisualize_PCA(
dataFilt = coadFilt,
dataDEGsFiltLevel = coadDEGsFiltLevel,
ntopgenes = 1000,
group1 = group1,
group2 = group2
)
</code>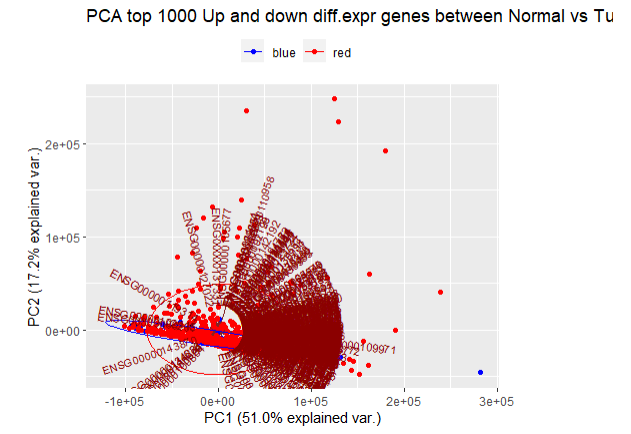
PCA
突变全景图
完全就是封装了maftools包,并且帮助文档里还没更新,还写着TCGAquery_maf,但是这个函数在最新版本的TCGAbiolinks里面已经没有了。。
maftools需要的文件如何自己整理
TCGA的maf突变文件不能下载了?直接用TCGAbiolinks包搞定!
<code style="margin-left:0">rm(list = ls()) # 加载突变数据 load(file = "./TCGA-SNP/TCGA-COAD_SNP.Rdata") coad.maf <- data </code>
直接画就行,和maftools一模一样,这里就不多介绍了,大家去用maftools吧。
<code style="margin-left:0">TCGAvisualize_oncoprint(mut = coad.maf,
genes = coad.maf$Hugo_Symbol[1:30]
)
</code>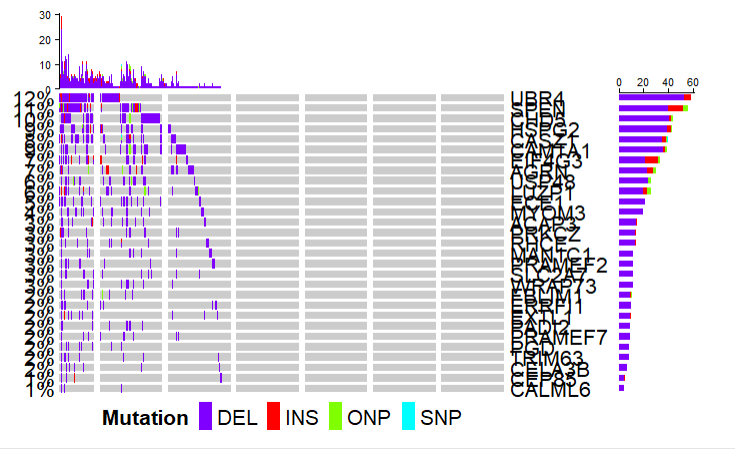
突变全景图
甲基化组间表达量/旭日图/条形图
可以参考之前的推文:
TCGAbiolinks的甲基化数据分析
新版TCGAbiolinks包学习:富集分析和生存分析
不得不说这些可视化函数有点鸡肋,不借助其他包是完全可以画出来图的,但是里面又都是封装的其他R包,而且还不如原装的R包好用!
未经允许不得转载:木盒主机 » 新版TCGAbiolinks包学习:可视化
 搬瓦工VPS最新优惠码 搬瓦工最高优惠6.81%优惠码 promo coupon code
搬瓦工VPS最新优惠码 搬瓦工最高优惠6.81%优惠码 promo coupon code  RackNerd:美国VPS 黑五优惠折扣 1核768RAM $10.28/年+神秘盒子 可随机减免金额
RackNerd:美国VPS 黑五优惠折扣 1核768RAM $10.28/年+神秘盒子 可随机减免金额 10G.BIZ【年终钜惠】美国/日本/韩国/香港独立服务器 秒杀仅24起,站群仅需99,三网CN2GIA五折抢购。CERA洛杉矶云服务器仅2.4起
10G.BIZ【年终钜惠】美国/日本/韩国/香港独立服务器 秒杀仅24起,站群仅需99,三网CN2GIA五折抢购。CERA洛杉矶云服务器仅2.4起 2022年RackNerd 美国VPS促销:4TB月流量11.88美元/年,支持支付宝,老优惠$9.89美元/年
2022年RackNerd 美国VPS促销:4TB月流量11.88美元/年,支持支付宝,老优惠$9.89美元/年